
“Benjamin Button disease” is a popular nickname for rare aging disorders in which a person appears much older or younger than their actual age. The term is inspired by the movie and story The Curious Case of Benjamin Button, but medically it is commonly linked to conditions like Progeria. This rare genetic disorder causes rapid aging in children and affects physical growth, skin, joints, and overall health. Although extremely rare, the condition has gained global attention for its unusual symptoms and emotional impact on families.
What Is Benjamin Button Disease?
You’ve probably heard the name from the 2008 Brad Pitt film. A man born old, aging in reverse, defying time. It’s a beautiful story, but the real Benjamin Button disease is something else entirely.
Benjamin Button disease is actually Hutchinson-Gilford Progeria Syndrome, a devastatingly rare genetic disorder that causes children to age at a dramatically accelerated rate. Instead of aging backward, children with progeria age forward fast. By the time a child with HGPS reaches age 10, their body often resembles that of a person in their 70s or 80s.
It affects roughly 1 in every 20 million births worldwide. At any given time, scientists estimate that fewer than 400 children worldwide live with classical progeria. In the United States, only a handful of cases are diagnosed each year.
The condition got its Hollywood nickname from the film, but the name “progeria” comes from the Greek word progeros, meaning “prematurely old.” It was first described by British physician Dr. Jonathan Hutchinson in 1886, more than a century before Brad Pitt ever stepped on set.
Read More: Robert Redford Disease
Meet Sammy Basso, The Real Face of Progeria
If you want to understand what Benjamin Button disease looks like in real life, start with Sammy Basso.
Sammy was the oldest person ever known to have lived with classic Progeria. Born in Italy in 1995, he was diagnosed at age two. Most children with the condition don’t survive past their mid-teens. Sammy lived to 28, nearly double the average lifespan.
But what made Sammy remarkable wasn’t just how long he lived. It was how he lived.
He earned a master’s degree in molecular biology with honors and went on to actively contribute to CRISPR/Cas9-based gene therapy research for the very disease that was slowly taking his life. He didn’t just survive progeria; he dedicated his life to understanding it, fighting it, and making sure the next generation of children would have better options than he did.
In a 2016 interview, Sammy said: “I like my life as it is, because it’s my life. I have my friends, my parents, and my family. These are the most important things. Progeria is a small part of my life, because it only affects the body.”
He collapsed and died due to suspected cardiovascular complications on October 5, 2024, at the age of 28. The world of science and the world in general stopped to mourn him.
Dr. Francis Collins, Sammy’s friend and research colleague, said, “We all knew that Sammy had a terrible circumstance that was not going to allow him to live a full life. But he was so vibrant. He was so alive. He was so engaged.”
His legacy didn’t end with his death. The gene therapy now being developed in his honor is called SamPro-2, named for Sammy himself. More on that shortly.
What Causes Benjamin Button Disease?
Here’s where it gets fascinating and a little mind-bending.
Every cell in your body has a nucleus, and inside that nucleus is a structure called the nuclear lamina, essentially the scaffolding that holds everything together. One of the key proteins that builds this scaffolding is Lamin A, encoded by the LMNA gene.
In children with progeria, there’s a single, tiny spelling error in that LMNA gene. One wrong letter in three billion. That’s all it takes.
That tiny error leads to a defective form of Lamin A called progerin
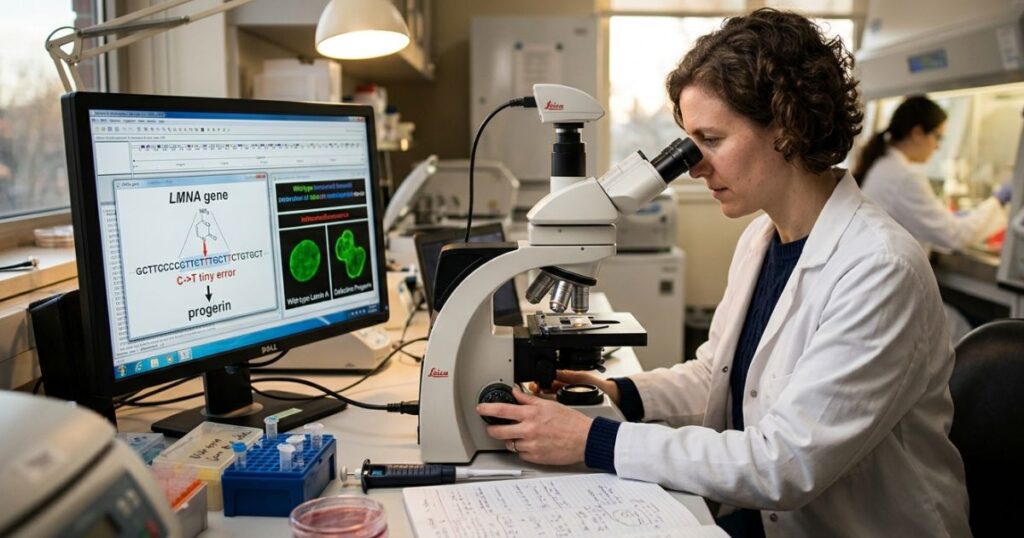
Progerin is toxic. It builds up inside cells, weakens the nuclear scaffolding, and causes cells to age and die far faster than they should. The result? A child’s body, skin, bones, heart, and blood vessels behave as if they belonged to a much older person.
Is Progeria Inherited? Can Parents Pass It On?
This surprises most people: progeria is almost never inherited from parents.
It’s what scientists call a de novo mutation, meaning it appears spontaneously, usually at the moment of conception. Both parents are typically healthy, with no family history of the condition. There’s currently no known way to predict or prevent it.
Your risk of having a child with progeria isn’t higher because of your age, ethnicity, diet, or lifestyle. It’s one of those extraordinarily rare events that just… happens. Which makes it one of the crueler twists of genetic biology.
Signs and Symptoms: What Does Progeria Look Like?
Babies with progeria are often born looking completely typical. The signs usually don’t appear until the first year or two of life, and then they arrive quickly.
Year-by-Year Symptom Timeline
Birth to 12 months:
- Normal appearance at birth in most cases
- Unusually tight, waxy skin may begin to develop.
- Slower than expected weight gain
Ages 1–2:
- Growth significantly slows in children with progeria, who are notably smaller and lighter than their peers.
- Loss of body fat and muscle, giving the face a distinctively aged appearance
- Hair loss (alopecia) begins, including eyebrows and eyelashes.
- Visible veins under the skin
Ages 3–5:
- The characteristic facial features become pronounced: a small face, a large head relative to body size, a beaked nose, and an underdeveloped jaw.
- Delayed or absent tooth development
- Stiff joints and hip dislocations become common.
- A high-pitched voice
Ages 6 and beyond:
- Atherosclerosis (hardening of the arteries) accelerates.
- Increased cardiovascular risk with each passing year
- Skin continues to age and tighten.
- Physical growth essentially stops.
The most important thing to understand about these symptoms is that the brain is not affected. Children with progeria have completely normal intelligence. They go to school, make friends, fall in love with subjects, and develop personalities and passions. Cognitively, they are entirely typical children, which makes the physical reality of the disease even more heartbreaking.
How Is Progeria Diagnosed?
If a doctor suspects a child may have Benjamin Button disease, the diagnostic process usually follows a clear path.
- Clinical observation: A pediatrician notices characteristic physical features, such as slowed growth, hair loss, aged skin, or an unusual facial structure. This typically happens between ages 1 and 2.
- Genetic testing: A blood test is taken, and the child’s DNA is analyzed. Doctors specifically look for the point mutation in the LMNA gene that causes progerin. This is the definitive confirmation.
- Cardiac evaluation. Because progeria affects the cardiovascular system, doctors will order heart tests, including echocardiograms and electrocardiograms, to assess arterial and cardiac function.
- Referral to the Progeria Research Foundation. The PRF, based in Boston, maintains the world’s only international registry of patients with progeria. Many families are connected to the PRF early in the diagnosis process, which opens access to clinical trials and global research networks.
Read More: Christina Applegate Disease
Can Progeria Be Detected Before Birth?
Currently, prenatal testing for progeria is not standard practice. Since the mutation almost always appears spontaneously rather than being inherited, there’s typically no reason to test for it in advance. However, if a family has had one child with progeria, genetic counselors may discuss testing options for future pregnancies.
How Long Do People With Benjamin Button Disease Live?
This is the question most people ask first, and it deserves a straight answer.
The average life expectancy for a child with classical progeria is 13–14 years. Without treatment, the leading cause of death is almost always cardiovascular disease, specifically heart attacks or strokes caused by advanced atherosclerosis. The same condition that affects adults in their 60s and 70s strikes these children decades earlier.
But averages tell only part of the story.
Sammy Basso lived to 28. Sam Berns, the American boy whose story inspired the documentary Life According to Sam, lived to 17. Some children live into their early 20s. The range is wide, and improving treatment options are pushing it wider every year.
What is the leading cause of death in progeria?
Atherosclerosis, the hardening and narrowing of the arteries, is the primary killer. The same toxic protein (progerin) that ages the skin and bones also devastates the walls of blood vessels. Over time, the arteries of a child with progeria begin to look like those of an 80-year-old, clogged and stiff. Eventually, the heart can no longer cope.
Is There a Cure for Benjamin Button Disease?
Here’s where the story gets genuinely hopeful, and science gets exciting.
Zokinvy (Lonafarnib): The First FDA-Approved Treatment
In November 2020, the U.S. Food and Drug Administration made history: it approved the first-ever drug for progeria, called Zokinvy (lonafarnib).
How does it work? Zokinvy belongs to a class of drugs called farnesyltransferase inhibitors. In simple terms, it blocks one of the steps that makes progerin so toxic, slowing the rate at which it accumulates in cells. It doesn’t cure the disease. But it buys time.
Read More: White Matter Disease Symptoms
With up to 11 years of clinical follow-up, lonafarnib-treated patients with HGPS had a survival benefit of 2.5 years compared with untreated patients.
Zokinvy reduced mortality by 72% and increased survival time by at least 4.3 years on average in patients with HGPS.
Those numbers might sound modest. But for a condition where every extra month matters, 2.5 additional years, and in some analyses, significantly more, is transformative. It means more birthdays. More school days. More time.
Gene Therapy: The Next Frontier
Zokinvy is a treatment, not a cure. The scientific community knows it. And they are working urgently on something bigger.
SamPro-2 is an experimental gene therapy currently in development, named in honor of Sammy Basso. It aims to go straight to the source: correcting or silencing the defective LMNA mutation using base editing technology, a precise form of gene editing that can change individual DNA “letters” without cutting the entire strand.
David R. Liu, the scientist leading base editing research, noted that Sammy himself recognized that a base editing treatment was among the most promising paths forward.
CRISPR-Cas9 approaches are also being explored, with promising results already seen in mouse models. Dr. Francis Collins confirmed that gene-editing experiments in mice with progeria have yielded promising results, with human clinical trials targeted for the near future.
Progerinin, a newer drug compound currently in Phase 2a clinical trials, is being tested in combination with lonafarnib. Early findings suggest it may work alongside Zokinvy to slow progerin accumulation through a different molecular mechanism, a potential one-two punch against the disease.
The pace of research right now is the fastest it has ever been. And every year, the children living with progeria today have better odds than the children who came before them.
Benjamin Button Disease vs. Normal Aging What’s the Difference?
At first glance, progeria appears to be accelerated aging. And in many ways, it is. But the two processes are not identical, and the distinction matters.
In normal aging, the body accumulates damage gradually over 70, 80, or 90 years. Cells slow down. Telomeres shorten. Tissues wear out. It’s a broad, system-wide process shaped by genetics, lifestyle, and environment.
In progeria, the aging process is driven by a single toxic protein, progerin. It’s targeted, molecular, and catastrophic. Children with the condition don’t experience every aspect of aging: they don’t develop Alzheimer’s disease, for instance, and their cognitive function remains entirely intact. What they experience is cardiovascular and structural aging at a devastating pace.
Here’s the remarkable twist: all of us produce tiny amounts of progerin as we age normally. Scientists have found that progerin accumulates in healthy adults over time, just at a rate too slow to cause illness in a typical lifespan. Studying progeria, then, isn’t just about helping 400 children. It’s about understanding the fundamental biology of how every human body ages. A cure for progeria could unlock insights that reshape how we treat heart disease and aging for millions of people.
The Progeria Research Foundation Fighting for Every Child
Behind every medical breakthrough in this story is one organization: the Progeria Research Foundation (PRF), based in Boston, Massachusetts.
Dr. Leslie Gordon co-founded the PRF in 1999 not only as a scientist but also as a mother. Her son Sam was diagnosed with progeria, and she refused to accept that nothing could be done. She went back to her lab and dedicated her career to changing the outcome for her son and every child like him.
Under her leadership, the PRF has:
- Identified the genetic mutation responsible for HGPS in 2003
- Launched the first clinical trials for lonafarnib at Boston Children’s Hospital in 2007
- Driven the FDA approval of Zokinvy in 2020, the first drug ever approved for the disease.
- Built the world’s only international progeria patient registry, connecting families and researchers across continents
- Initiated gene therapy research that is now advancing toward human clinical trials
Read More: What Disease Does Sam Elliott Have
The PRF described Sammy Basso as someone who “left behind a legacy of laughter, hope, and a passionate drive to advance the science of Progeria.” That describes the foundation’s spirit, too.
If you want to support their work, visit progeriaresearch.org. Every dollar funds research that genuinely and measurably extends children’s lives.
Conclusion
Benjamin Button disease, often associated with Progeria, is a rare condition that causes accelerated aging and serious health complications. While there is currently no complete cure, early diagnosis, supportive treatment, and medical care can help improve quality of life. Greater awareness and research continue to bring hope for better treatments in the future.
FAQs
What is the real name for Benjamin Button disease?
The medical name is Hutchinson-Gilford Progeria Syndrome (HGPS). It’s named after Dr. Jonathan Hutchinson, who first described the condition in 1886, and Dr. Hastings Gilford, who studied it further in 1904. The “Benjamin Button” nickname became widely used after the 2008 Brad Pitt film.
How rare is Benjamin Button disease?
Extraordinarily rare. It affects approximately 1 in 20 million children worldwide. Scientists estimate that at any given time, fewer than 400 children worldwide are living with classical progeria. In the United States, only a small number of new cases are identified each year.
Is Benjamin Button disease contagious?
Not at all. Progeria is a genetic disorder caused by a spontaneous mutation in the LMNA gene. It cannot be caught, transmitted, or spread from one person to another.
Does progeria affect intelligence or the brain?
No. This is one of the most important facts to understand. Children with progeria have completely normal intelligence and brain function. The condition affects the body’s physical structure, skin, bones, heart, and blood vessels, but leaves cognitive ability entirely intact. These are bright, curious, fully aware children.
What drug treats Benjamin Button disease?
The only FDA-approved treatment is Zokinvy (lonafarnib), approved in November 2020. It’s a daily oral medication that slows the toxic effects of the progerin protein, extending life by an average of 2.5 years in clinical trials. It is not a cure, but it meaningfully improves survival odds.
Can someone with progeria have children?
In theory, it’s biologically possible, but in practice, the condition’s shortened lifespan means most children with progeria do not survive to typical reproductive age. There are no recorded cases of a person with classical HGPS having biological children.